

The title of this post might seem an elementary question, and one for which a satisfactory answer would appear in an Internet search within a few seconds after clicking the mouse. However, I believe that that this is one of those questions for which many people believe mistakenly, that scientists know the answer. It is also a question that I am asked time and time again, and so I thought I would provide my own short perspective on the “known knowns, the known unknowns, the unknown knowns ….and the unknown, unknowns” of how enzymes work. In short, while there are some general principles that apply to all enzymes, each of the many thousands of naturally occurring enzymes holds its own intrinsic, scientific secrets.
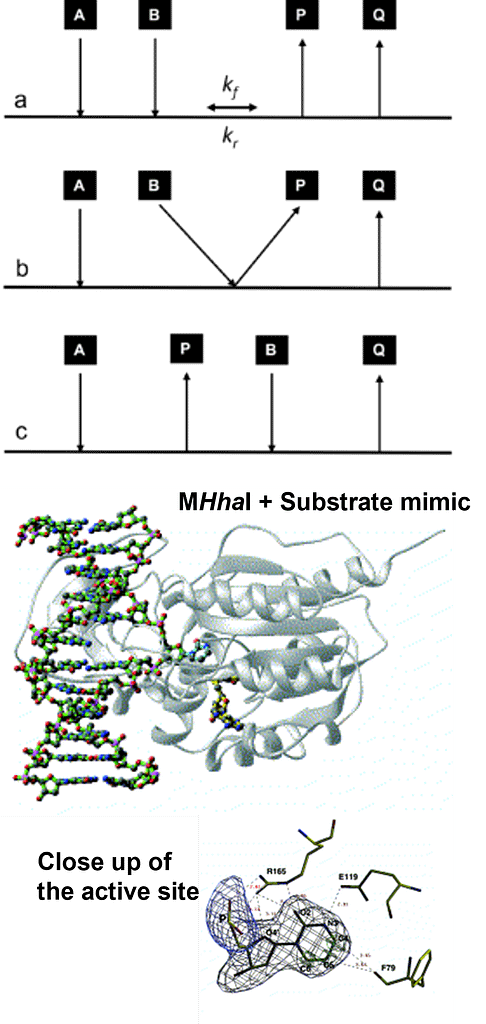
To whet your appetite, the enzyme shown, M.HhaI, recognises a stretch of double-helical DNA containing the sequence GCGC (purple), and transfers a methyl group from a donor molecule (yellow) to the internal cytosine ring (green): in doing so, the enzyme (orange) first plucks the base out of the Watson-Crick helix, methylates it; only letting go at the end of the reaction.
If, like me you find such phenomena truly incredible, then read on…
Today’s students are introduced to enzymes as biological catalysts in high school. However, I suspect that on hearing the word enzyme, most people think of biological washing powders. Simply put, enzymes are molecules, found in Nature, that speed up chemical reactions under a broad range of physiological conditions. Apart from their essential role in sustaining life, where they represent about 10% of the information encoded in our chromosomes, enzyme extracts have been harnessed for hundreds of years by bakers, cheese makers, brewers and more recently, by the pharmaceutical industry, where enzymes are used not only to facilitate drug synthesis, but as therapeutic agents in their own right.
Let me begin with a list of key words and abbreviations, commonly used by enzymologists, to help with this discussion.
Catalyst, catalysis and catalytic. These terms are derived from a Greek word that means to dissolve. It was formalised into chemistry by Berzelius around 200 years ago to describe the properties of a chemical substance that stimulates a chemical reaction but remains unchanged at the end of that reaction.
Active site. This is a highly organised location (often called a cleft) within an enzyme, that usually occupies between 10 and 40% of the volume of an enzyme molecule, where catalysis occurs.
Substrates (or reactants) and products. Each enzyme catalyses a chemical reaction involving one or more molecules. The molecule(s) entering the active site are referred to as substrates (or reactants), while the molecule(s) leaving the active site, after a reaction has taken place, are called products.
Primary, secondary, tertiary, and quaternary structure. Most naturally occurring enzymes are unbranched polymers of amino acids (polypeptides), linked via a peptide bond having a unique sequence of amino acids: this is referred to as the primary structure of an enzyme (or indeed any protein). In vivo, the primary structure of an enzyme emerges from the ribosome during protein biosynthesis and immediately begins to form intramolecular interactions, driven by the general tendency for hydrophobic amino acid side chains to associate with each other, and for hydrophilic amino acids to engage with the aqueous environment in the cell.
In the seconds taken for the biosynthesis of the complete primary structure to occur, the fluctuating intramolecular interactions often lead to the formation of regular geometric elements that include α-helices and β-sheets, interspersed with less regular stretches of polypeptide chain. These are termed secondary structures and are stabilised by regular interactions that include hydrogen bonds, between the carbonyl oxygen of one amino acid, and the amino hydrogen of another.
The tertiary structure is the unique three-dimensional arrangement of atoms of an enzyme: it is determined by the primary structure under physiological conditions. Many years of experimental structure determination, using X-ray diffraction, high resolution NMR spectroscopy and more recently, cryo-electron microscopy (EM), have shown that proteins adopt their structures with differing degrees of stability and some proteins possess unstructured regions that often respond to the addition of one or more ligands. In some extreme circumstances, some proteins are naturally unstructured, as part of their biological role. In many situations, one or more tertiary structures combine to form quaternary structures: these units are often called subunits or protomers. Such macromolecular assemblies often form spontaneously, immediately after polypeptide chain synthesis, but in other situations there is a more dynamic exchange of subunits as part of their physiological function.
Ribozymes. Not all, but most, naturally occurring enzymes are polypeptides. Around 40 years ago, Tom Cech and Sidney Altman independently reported that certain RNA species exhibited catalytic behaviour which, until then, had been limited to proteins. This discovery opened the floodgates for a new field of research, important for our understanding of the origins of life on earth, and fundamental aspects of enzymology. In addition, the development of a wide range of technologies associated with the synthesis and purification of RNA, ultimately underpinned the development of mRNA vaccines, that have been so critical in limiting the recent pandemic. Ribozymes, or catalytic RNAs, occupy biological niches, in evolutionary terms, but just like the side chains of amino acids in proteins, there is no a priori reason why the functional groups of nucleotides, appropriately configured in space, cannot perform a similar role in converting substrate molecules to products. In fact, the active site of the “mega” enzyme, the ribosome, that catalyses protein synthesis, is itself a large complex of RNA and protein molecules: with RNA providing the key catalytic functions. I shall leave further discussion of the fascinating properties and applications of ribozymes for another time.
Kinetic parameters: (Vmax, kcat and Km). These are the terms used to provide the metrics associated with the catalytic properties of a particular enzyme. Generally speaking, a user of enzymes needs to know the maximum rate achievable by a given quantity of enzyme under a specified set of conditions. The maximum velocity (Vmax) of a given amount of an enzyme at a fixed pH, temperature and at a specified substrate concentration will determine is probably the most important “metric” for industrial applications, in which an enzyme is added to “process” a substrate (for example). The amount of enzyme is often supplied in mg quantities and if “pure”, this information can be used to calculate the number of moles in a given sample. Vmax is usually expressed as the number of moles of substrate converted to product per mg enzyme, per minute (or second). This is sometimes converted to kcat, which has units of reciprocal time, since the concentrations of both substrate and enzyme are expressed as molarities. Finally, the term Km , is often used as a shorthand to define the strength of interaction between an enzyme and its substrate, which provides insight into the specificity of that enzyme for its substrate: a substrate with a low Km, has a higher affinity for the active site than one with a high Km. This definition arises since under certain conditions, the Km is equal to equilibrium dissociation constant for the enzyme substrate interaction and has units of molarity.
Transition state. The chemical pathway from substrate to product, is usually bracketed by two thermodynamically stable states. For example, both the polypeptide chain (the substrate) and the peptides (products) released by the action of the protease trypsin, are both relatively stable molecules. It has been suggested that the active site of all enzymes has a preference for binding and transiently stabilising a molecular form of the substrate that in simple terms represents a form that is mid-way between substrate and product, referred to as the transition state. The nature of any transition state, by definition a fleeting molecular entity, makes it challenging to observe. One piece of evidence for this central tenet of enzyme catalysis, is that antibodies raised against transition state mimics, act as catalysts when presented with the ground-state substrate. Suffice to say, the preferential stabilisation by an enzyme of the transition state, is a key element underpinning enzyme catalysis.
I should add a note of caution here. The kinetic analysis of enzyme catalysed reactions is a complex process in which variations in initial rates of reaction result from a range of factors from temperature and pH to the concentrations of the substrates and products. Moreover, depending on the mechanism of the reaction, parameters such as Km can have complex origins. In addition, not all enzymes follow the “rules” established by early pioneers: the variation of initial rates may depend on ligand-induced enzyme conformational equilibria, thereby requiring more sophisticated forms of kinetic analysis; included in this group are the regulatory, or allosteric enzymes, which warrant a separate post. There are many excellent textbooks covering enzyme kinetics and mechanism, but I recommend the classic text by Alan Fersht entitled “Enzyme Structure and Mechanism” which can be picked up second hand (any edition will provide an excellent place to start), and the recent excellent monograph on enzymes by Paul Engel.
I think it is fair to say that many people feel comfortable using the term enzyme, but I am not so sure that they appreciate how challenging it has been to determine just how enzymes work at the molecular level. From the earliest attempts to formalise the mechanistic analysis of enzymes around 125 years ago, through to the determination of many enzyme structures in atomic detail, often with substrates bound to the active site, many aspects of the question posed by this post, remain unresolved. Here, I shall take a critical look at how our understanding has evolved since their discovery at the end of the nineteenth century, focusing on the general principles underpinning enzyme catalysis, with a few examples.
JBS Haldane’s landmark book on Enzymes, written over 90 years ago, makes for fascinating reading today, and while many of the most enduring discoveries in our understanding of enzymes were to follow; I believe it is instructive to revisit some of the fundamental observations that underpin our understanding of what enzymes do, before we consider how they do it.

One of my favourite demonstrations of a transition-metal-catalysed reaction, is the enhanced decomposition of hydrogen peroxide by compounds, such as powdered manganese dioxide. Within a few seconds of adding the fine, black powder to a beaker containing hydrogen peroxide, bubbles of oxygen appear, the temperature begins to rise, and within a few minutes, decomposition is so fierce that steam is evolved from the reaction vessel. In living organisms, the enzyme catalase catalyses the decomposition of hydrogen peroxide, although a few microbes that lack genes encoding catalase, eliminating the problems associated with dangerous levels of intracellular hydrogen peroxide, by pumping in manganese ions. Importantly, however, the exothermic nature of manganese-mediated decomposition is incompatible with physiological temperature control. Physiological temperatures typically range from just above zero to just below 100°C, where water remains liquid. There are exceptions to this rule among the so-called extremophiles, but I will reserve discussion of this fascinating physiological topic for another post. Remarkably, Catalases are capable of decomposing (technically disproportionating) many tens of millions of H2O2 molecules per second
Perhaps the first question that a biochemist should consider, is whether we can learn anything from studying chemical catalysis, to help us understand how enzymes work? As mentioned above, in Nature, examples exist of organisms that harness the power of manganese ions to catalyse hydrogen peroxide decomposition, but most organisms deploy the enzyme catalase to remove any unwanted hydrogen peroxide that accumulates as a by-product of intracellular metabolism.
A second question arises in my mind: catalase enzymes comprise one or more polypeptide chains, supporting a metal ion such as iron or manganese at the active site (which we shall come onto shortly), and therefore represent a considerable investment in energy for protein synthesis. Manganese ions, on the other hand can be extracted from the external environment, but this usually requires the help of a dedicated membrane-bound polypeptide: a manganese-specific transporter. So the energetic cost of the two solutions, may not be that different.
Next, I might consider whether enzymes are any “better” than inorganic catalysts at speeding up a chemical reaction? The (relatively) simple atomic structure of a solvated transition metal catalyst is in stark contrast to the complexity of an enzyme like catalase. If we are to understand enzymes as catalysts, we need a standardised methodology for measuring reaction rates and an agreed set of “units” for measuring the rate of catalysis. Calculations suggest that catalases are over 100 times more efficient at catalysisng peroxide decomposition than metal ions like manganese. I believe there is always value to be had in comparing inorganic catalysts with simple organic catalysts and enzymes, since the simplicity of measurements with salts and small molecules facilitates precise measurements, but ultimately, in order to understand enzyme mediated catalysis, the enzymatic reaction must be analysed and considered in its own right.
Let me next provide a short introduction to enzyme metrics. There are two ways of capturing the rate of a reaction. The most common method involves quantifying the amount of product(s) formed just after a reaction is initiated. In the example above, hydrogen peroxide decomposition is typically measured as a function of the volume of oxygen liberated over a given time. It is also possible to measure the consumption of one or more reactants: in the case of a reaction in which NADH is consumed, there is an accompanying decrease in absorbance measured at a wavelength of 340nm. Both approaches are widely used and are generally selected based on the experimental simplicity and reproducibility of the detection method.

The rate of reaction is expressed in several ways, but essentially comprises units of concentration measured over time. For example, the rate of accumulation of product (or disappearance of substrate), may be expressed as mmol.min-1, or as µmol.s-1: whatever the units, it is vital that when comparing reaction rates, the units are normalised with respect to both concentration and elapsed time. The measurement of the rates of enzyme catalysed reactions typically requires very small quantities of enzyme and primarily for this reason, enzyme kinetics became a very popular experimental (and theoretical) field for biochemists, in the middle of the last century. In fact, some of the most significant advances in the field, were made prior to our ability to visualise enzymes interacting with their substrates in molecular detail.
One of the challenges of enzymology has always been the reliable determination of substrate, product and especially enzyme concentration. When purifying, or comparing enzyme activity, measurement of the “specific activity” of an enzyme preparation is critical. This refers to the number of units of activity per mole of an enzyme. The specific activity of any enzyme should be carefully monitored during its preparation and purification; moreover, care should be taken to store an enzyme preparation under conditions were the loss of specific activity is kept to a minimum.
From the incredibly prescient ideas in the 1890s, about how enzymes and substrates combined, by the Nobel Laureate, Emil Fischer, to the first crystallographic images of the enzyme lysozyme engaging with its carbohydrate substrate in its active site (David Phillips and his group at Oxford in the 1960s): the experimental and theoretical field of enzyme kinetics, led to the development of highly sensitive methods for measuring enzyme catalysis, by pioneers including Michaelis and Menten, Britton Chance, Daniel Koshland and the multi-disciplinary group from France of Monod-Wyman-Changeux (and many more, too numerous to mention here). In fact, some of the most significant advances in the field, were made prior to our ability to visualise enzymes interacting with their substrates in molecular detail.
The diagrams on the right, provide a schematic illustration of substrates (A and B) binding to the active site of an enzyme (depicted by a horizontal line) followed by the release of products (P and Q). Some enzymes catalyse reactions involving a single substrate and a single product, but most reactions involve two or more substrates and products. The examples given are for a two substrate:two product enzyme catalysed reaction. The order of addition of A and B in (a) is ordered and the release of products P and Q is also ordered. The reaction (as with all enzyme-catalysed reactions) is reversible, and kf and kr represent the rates of the forward and reverse reactions respectively. This representation of enzyme reactions allows for the simple description of reactions, such as that shown in (b), where the addition of the second substrate (B) leads to the spontaneous formation of the first product (P). This is referred to as a Theorell-Chance mechanism, after its discoverers: there is no significant formation of a ternary complex (Enzyme + A +B). The scheme shown in (c), represents the addition of one substrate (A), followed by the release of the product, P. In a second phase, substrate B enters the active site and finally product P is released. This is sometimes called a “ping-pong” reaction and is common among group transfer reactions, in which a chemical group is extracted from the first substrate and transferred to the second. I personally find this simple notation very useful when explaining the sequence of events during an enzyme catalysed reaction, without the need to incorporate structural information.
The images below add a structural element to understanding enzyme mechanism. The upper image on the right shows a complex between the enzyme M.HhaI and a tight binding nucleic acid substrate, along with the methyl donor, S-adenosyl-L-methionine. The close up below, shows how a constellation of conserved, active site amino acid side chains bring specificity to the enzyme substrate interaction. (The original publication can be located here). This is a general theme in enzyme catalysis.
The home straight….One way of addressing the question posed in this Blog, is to consider the differences between enzymes and the genes that encode them. Both are polymers, and while the vast majority of enzymes are unbranched chains of amino acids, some, such as the hammerhead ribozyme, are ribonucleic acids. Deoxyribonucleic acids have largely evolved to store and transfer information, and as such they adopt a stable double helical structure, transiently separating and unwinding during replication, transcription, repair, and recombination. DNA largely gains its topological complexity from its interactions with proteins. Ribozymes, are single stranded polymers of RNA, often associated with proteins, but where a defined 3D structure arises through the interplay between intra-molecular double helical segments, often flanked by loops, and stabilised by metal ions, in particular magnesium ions.
Protein based enzymes, as discussed earlier, adopt stable 3D structures, a feature that is largely hard-wired into the sequence of amino acids (recall, the primary structure), that spontaneously folds into a unique 3D structure. As always there are caveats and exceptions, but I shall concentrate here on the generalities and assume that all protein enzymes follow the principles associated with Nobel Laureate Christian Anfinsen. The important message here is that the enzymes are genetically encoded and that a bacterium like E. coli, which possesses the capacity to express over one thousand enzymes, does so with a wide dynamic range (some enzymes are expressed in a handful of copies and some in the tens of thousands) completes its life in around 30 minutes. It is clear that the enzymes required to fuel the cell and build it up, must do so with incredible efficiency. DNA, in contrast is a single copy macromolecule, replicated once during this 30-minute period.
Following synthesis on the ribosome, each enzyme folds spontaneously and a catalytically competent molecule encounters its substrate(s) through general diffusion, at which point it fulfils its physiological function. The complementarity between the structure of the active site of the folded enzyme (which may represent less than 10% of the total molecular volume), and the transition state of the substrate(s) provides the substrate with a favourable pathway to product formation, and simultaneously provides the selectivity associated with most enzymes.
The general view is that enzymes achieve significant rate enhancement through preferential stabilisation of the transition state over the ground state of a substrate. Of course, the details of how this is achieved is specific to each enzyme, although the physico-chemical solutions to this are often characterised by conservation of the amino acids responsible for these interactions within the active site of the enzyme. At Entropix, the evolutionary tolerance to amino acid substitution exhibited by different classes of enzymes, throughout their primary structures, encoded by generations of enzyme variants, provides us with valuable insight during our enzyme development programmes.
I like to think of the enzyme hanging on to the transition state in the same way that one hangs onto a bar of wet soap. The grip should be firm, but at the same time, the slightest movement may cause the soap to shoot out of your hands. During this moment, those amino acid side chains, and in some instances, reactive species from a cofactor or metal ion (see my last Blog), approach the transition state and promote the formation of products, which are then released. The enzyme is then ready to accommodate a second, similarly oriented reactant molecule, and so forth. The rate limiting step (the slowest step) of an enzyme catalysed reaction can be associated with the chemistry of the reaction, or often the release of reaction products, which may be closer to the structure of the transitions state, than the substrate. While methods are widely available to investigate these issues, importantly, it is through directed evolution that variants arise in vitro, that overcome barriers to efficient catalysis: barriers that may well be irrelevant in the organism from which the enzyme is derived.
One of other key features about all catalysts is that they do not change the intrinsic equilibrium position of a specific reaction. Rather, they simply provide a route to overcoming the kinetic barriers, necessary for the chemical conversions to take place on a physiological timescale. There is no magic, just molecular strategies that persuade otherwise stable molecules to react at neutral pH and at blood heat (in most cases). This is in stark contrast to the temperatures, pressures, and extremes of pH, required for many historic manufacturing processes. In addition, the non-equilibrium nature of life, means that enzymatically derived products, generally form the basis for a downstream enzyme-catalysed reaction. This metabolic flux serves to pull the equilibrium over to the right, which in turn, enables metabolic pathways like glycolysis and protein synthesis meet their physiological deadlines with time to spare.
The detailed events that take place in the active site of an enzyme, follow the rules of mechanistic organic chemistry. Again, there is no magic, but rather, typically stable compounds like glucose, fatty acids and amino acids and the biopolymers that include nucleic acids and proteins, are persuaded to ligate, isomerise, hydrolyse etc., when induced to adopt a transient, unstable configuration, in close proximity to a constellation of a small set of amino acid side chains, for less than one second.
With this general description of enzyme catalysis in hand, what features are generally associated with all naturally occurring enzymes?
Molecular weight. Enzymes typically have subunit molecular weights of between 15kDa and 200kDa. As always in Nature, there are some higher molecular weight enzymes, but most enzymes are typically around 50kDa. Many enzymes are oligomeric: some are homo-dimers (2 copies of the same polypeptide chain), some are homo-tetramers and some homo-pentamers (there are many arrangements found in Nature). Some other enzymes are hetero-oligomers, with one or more copies of different polypeptide chains coming together to form the active enzyme species. The latter class includes complex, highly regulated enzymes such as those involved in nucleic acid biosynthesis and energy transduction (as just two of many examples).
pH and temperature optima. All enzymes exhibit some form of optimum temperature: it may not always correspond with the physiological body temperature, but the enzyme will have a level of activity and stability sufficient to meet the metabolic needs of the organism at its normal growth temperature. The catalytic activity of an enzyme is also usually characterised by a pH optimum, often described by a bell-shaped curve (although not always mathematically ideal). This is a result of the influence of ionisable amino acids in maintaining the stability of the folded state of the enzyme, and the chemical reactivity of amino acid side chains in the active site. While in some cases temperature and pH optima are described by bell-shaped curves, in many cases there is a much less idealised relationship.
Reaction rates. What is the range of reaction rate enhancements that are catalysed by enzymes? This is a tough one. Enzymes in Nature have evolved through mutational events, combined with natural selection over evolutionary time. In directed evolution, these processes are experimentally compressed into days and weeks. However, not all reactions are chemically equivalent, in fact there are considerable levels of metabolite diversity in Nature. Some enzymes are only capable of a single turn-over event (technically these are not catalysts), but enzymes like catalase can crank through substrate molecules at rates that appear to cofound the laws of chemistry. In other words, some reactions are catalysed by several-fold and some by many thousand-fold. In order for a catalytic reaction to take place, the substrate has to make a productive collision in solution (usually water) with an enzyme molecule. Many collisions are non-productive, and diffusion of molecules is a function of molecular volume and shape (in the case of polymers). The portfolio of enzymes present in say, a one litre culture of E.coli, has the capability of catalysing the conversion of 10mM glucose from a single colony of E.coli, overnight into around 5g cell paste. Collectively these enzymes operate as a proteome, with some enzymes producing 10-100-fold enhancement and some a million-fold rate enhancement. Of course, the slowest reactions in central metabolism (the rate limiting steps), ultimately “call the shots”.
The prospect for enhancing the catalytic performance of enzymes remains uncharted territory, but it is clear that even the slowest of enzymes can be enhanced by a programme of directed evolution, which can draw on selective pressures that are often incompatible with whole organism survival. I realise that this post may seem somewhat verbose, but in fact the published material on enzymes over the last hundred years is truly vast. I have only touched the surface of this body of knowledge, but I hope in part 1, I have helped provide some insight and whetted your appetite for learning more about this fascinating area. In the second part of this post, I shall look at some classic examples from the literature that have been influential in our understanding of the principles of enzyme catalysis, and the questions that remain unanswered.